Comparative metagenomics
Alejandra Escobar 
Christian Atallah
Normalization methods, alpha & beta diversity, and differentially abundant features
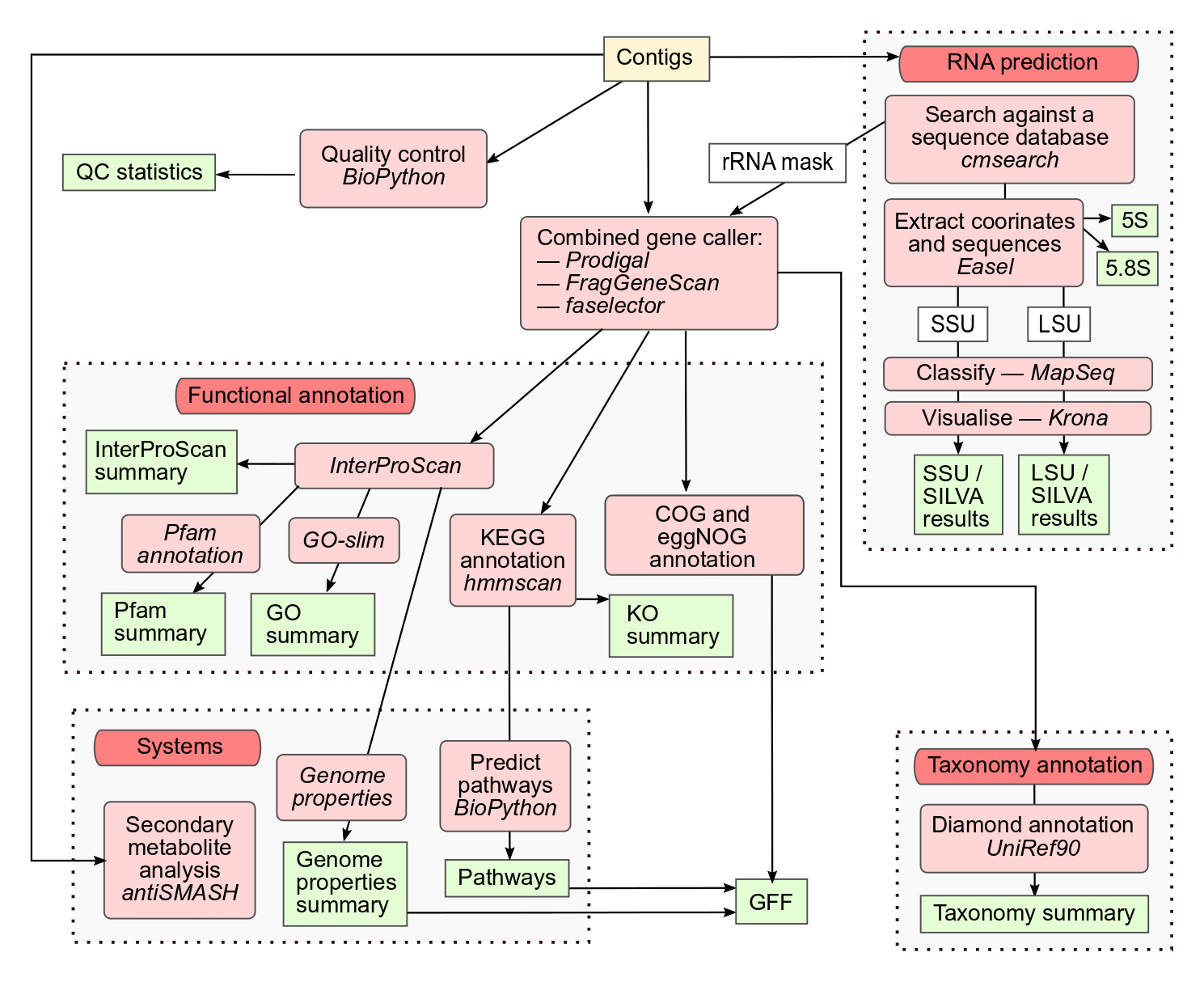
In this practical session, we aim to demonstrate how the MGnifyR tool can be used to fetch data and metadata of a MGnify metagenomic analysis. Then we show diversity metrics calculus and two methods to identify differentially abundant features using taxonomic and functional profiles generated through the MGnify v5.0 pipeline for metagenomic assemblies, as shown in the workflow schema below.
The dataset is the TARA ocean metagenomic study (WMS) corresponding to size fractions for prokaryotes MGYS00002008. Find more information about the TARA Ocean Project.
MGnifyR is a library that provides a set of tools for easily accessing and processing MGnify data in R, making queries to MGnify databases through the MGnify API. The benefits of MGnifyR are that data can either be fetched in TSV format or be directly combined in a phyloseq object to run an analysis in a custom workflow.
The exercises are organized into 5 main sections:
- Fetching data and preprocessing
- Normalization, alpha diversity indices and taxonomic profiles visualization
- Comparative metagenomics at community level: Beta diversity
- Detection of differentially abundant taxa (SIAMCAT)
- Detection of differentially abundant functions (Aldex2)
Open the Jupyter Notebook
The practice has been prepared in a Jupyter Notebook available in the MGnify Notebooks Github repo.
Yellow boxes:
- Up to you: To re-run the analysis changing parameters
- Questions: For open discussion
Blue boxes:
- Notes: Informative boxes about the running command
- Tips: Information useful for results interpretation
Use the Jupyter Notebook after the course
This notebook is based on a publicly accessible version. You can use this at any time.
- It is available to use from your web browser, no installation needed: notebooks.mgnify.org
- You can see a completed version of it, with all the outputs, on docs.mgnify.org
- You can use a prebuilt docker image and our public
notebooksrepository: github.com/ebi-metagenomics/notebooks. This should work on any computer you can install Docker on. - You can try and install all the dependencies yourself
¯\_(ツ)_/¯